
 How to resolve AdBlock issue?
How to resolve AdBlock issue? 
Artículos
Las matemáticas de la Química
Investigadores del Grupo de Sistemas Complejos de la Universidad Politécnica de Madrid consiguen explicar la compleja dinámica vibracional de sistemas moleculares utilizando métodos geométricos
Pese a lo temidas que han sido siempre las matemáticas, hoy en día son, sin lugar a dudas, una de las carreras de moda. Este éxito actual tiene su origen, sobre todo, en el boom que hemos visto a lo largo de los últimos años con el desarrollo del big data y la inteligencia artificial, aunque también se debe a que se ha puesto de manifiesto su importancia en otras muchas disciplinas. Así, sin las matemáticas no seríamos capaces de diseñar puentes, fabricar aviones, crear modelos en ecología, analizar redes sociales…
La lista puede ser prácticamente infinita y la química no está fuera de ella. De hecho, a lo largo de los últimos años, numerosos investigadores han comenzado a estudiar el comportamiento de sistemas químicos usando métodos geométricos de la llamada teoría de los sistemas dinámicos de las matemáticas.
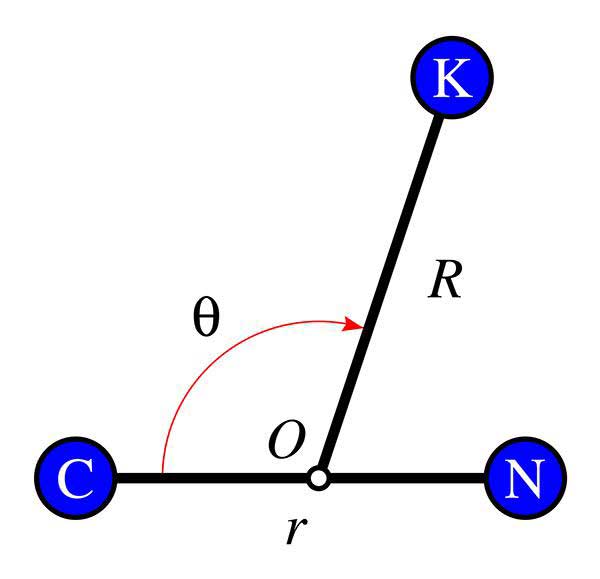
En un reciente artículo publicado en la revista Communications in Nonlinear Science and Numerical Simulation, investigadores de la Universidad Politécnica de Madrid (UPM)ꟷen colaboración con la Universidad Autónoma (UAM) y el Instituto de Ciencias Matemáticas (ICMAT)ꟷ han aplicado los descriptores lagrangianos de las matemáticas a la molécula de cianuro de potasio (KCN), y han conseguido explicar y reproducir su compleja dinámica vibracional incluso a altas energías, límite donde los métodos más tradicionales no tienen validez.
Los descriptores lagrangianos son unos indicadores desarrollados originariamente para el estudio de la dinámica de los océanos que permiten identificar las estructuras geométricas (llamadas variedades invariantes) y que determinan cómo cambia con el tiempo un sistema (molecular, en este caso).
Lo interesante de esta herramienta es que para usarla es, básicamente, sólo necesario integrar las ecuaciones del movimiento, sin necesidad de llevar a cabo complejos estudios de la dinámica cercana a las trayectorias, como ocurre con los indicadores de caos tradicionales, lo que facilita mucho el estudio. Los resultados del trabajo servirán para el desarrollo de una química selectiva en la que la energía vibracional puede contribuir a facilitar reacciones que de otro modo no se llevarían a cabo por la alta energía translacional que sería necesaria.
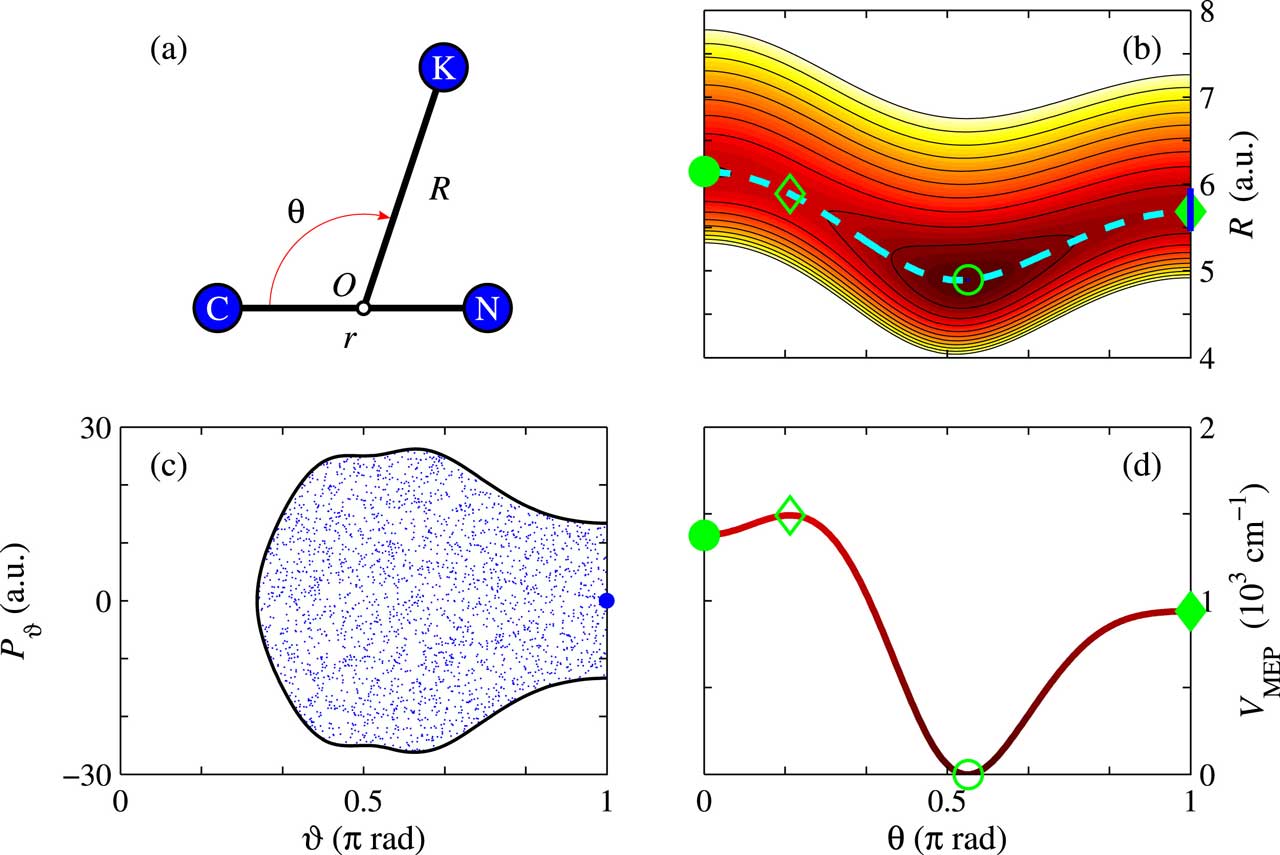
Sistema molecular KCN estudiado, formado por un átomo de potasio (K), un átomo de carbono (C) y un átomo de nitrógeno (N), y coordenadas (R, θ, r) que describen la configuración de la molécula. Fuente: UPM.
En el trabajo publicado, en el que han participado investigadores del Grupo de Sistemas Complejos de la UPM, se ha comprobado que las estructuras geométricas (variedades invariantes) asociadas a una única órbita periódica del KCN determinan cómo cambia de configuración toda la molécula haciendo que los átomos que la forman vibren de una forma específica. Según declara Fabio Revuelta, investigador de la UPM participante en el estudio: “El conocimiento de estas vibraciones es muy importante para el desarrollo de una química selectiva, en la que la energía (vibracional) puede contribuir a facilitar reacciones que en otro caso no se llevarían a cabo por la alta energía (translacional) que se necesitaría. Además, aunque a baja energía existen otros métodos para el estudio de las vibraciones en sistemas moleculares, por ejemplo, el análisis de modos normales, estos métodos no resultan válidos a energías altas como a las que se ha realizado el estudio, de ahí el interés de nuestro trabajo”, añade el investigador.
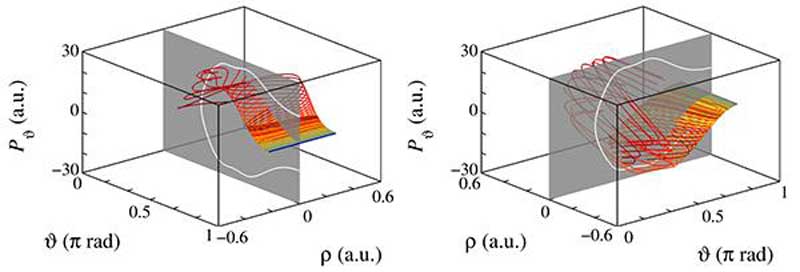
Las variedades invariantes (inestable a la izquierda y estable a la derecha) asociadas a la órbita vibracional que determinan la dinámica de la molécula de KCN aparecen como segmentos rectos cerca de la órbita de referencia, pero luego se pliegan y deforman, dando lugar a una dinámica fuertemente caótica. Fuente: UPM.
Finalmente, los investigadores también observaron que las variedades invariantes estudiadas tienen un comportamiento mucho más complejo del esperado, plegándose y enrollándose sobre sí mismas de una forma más abrupta que en otras moléculas. Estos plegamientos son los responsables de que la dinámica vibracional de la molécula de KCN sea mucho más caótica que en otras moléculas formadas también por tres átomos. Esto se debe a que las interacciones entre sus constituyentes son mucho más complejas en este caso, aunque ꟷconcluye Fabio Revueltaꟷ: “eso ya es otra historia que requiere, de nuevo, de más matemáticas”.
- Revuelta, F. J. Arranz, R. M. Benito, F. Borondo, Unraveling the highly nonlinear dynamics of KCN molecular system using Lagrangian descriptors, Comm. Nonlin. Sci. Num. Sim. 123, 107265 (2023).
- Revuelta, R. M. Benito, F. Borondo, Identification of the invariant manifolds of the LiCN molecule using Lagrangian descriptors, Phys. Rev. E 104, 044210 (2021)
- Revuelta, R. M. Benito, F. Borondo, Unveiling the chaotic structure in phase space of molecular systems using Lagrangian descriptors Phys. Rev. E 99, 032221 (2019)







